In our previous Blog, we explored the importance and applications of RNA-Seq for plant transcriptomics and crop sciences. Even though analyzing a plants transcriptome can shed light on regulatory mechanisms, gene expression in response to stressors or expression networks during plant growth and development, researchers have to face several unique challenges when working with plant samples, starting with RNA extraction. In this article we will describe the specific challenges for RNA isolation from plant material, highlight commonly used methods, and give tips for efficient and successful extraction of high quality RNA. For a general overview about RNA Extraction and RNA quality control prior to NGS experiments, check out our dedicated chapter in the RNA Lexicon.

Challenges for Plant RNA Extraction and Tips to Improve Yield and Purity
Plants have several characteristics that can impede extraction of high-quality RNA. First of all, plants possess a rigid cell wall that needs to be disrupted to allow efficient extraction. In contrast to animal cells which can be simply transferred to lysis buffer for RNA extraction, plant cells require mechanical or enzymatic disruption. Researchers therefore often flash freeze plant tissues in liquid nitrogen and grind the frozen tissue with pestle and mortar to a fine powder that can be extracted. In addition to the cell wall, the outer surface may also be coated with wax, as for example the needles from evergreen trees or it can contain fibrous lignin tissue, e.g., wood or bark. Harsh mechanical disruption, which is required to physically break plant cells, can affect the integrity of the isolated RNA (or DNA).
As a second factor, RNases can be present in higher amounts in some plant species leading to RNA degradation during harvest or the extraction procedure. Inactivating RNases immediately is therefore critical to obtaining high-quality intact RNA. The use of extraction buffers which efficiently inactivate RNases can help preserve integrity.
Tipp: Samples should be flash frozen and kept cold during the disruption process to keep RNases in an inactive state. Consider also to keep lysis or isolation buffers cold (+4 °C or on ice) and to perform lysate clearing steps in pre-cooled centrifuges.
As various plant tissues contain a high water-content, the quantity of RNA that can be extracted from these is often quite low. To process the low concentrated RNA, researchers either need highly sensitive downstream assays that can cope with low input amounts, or they need to perform concentration steps, e.g., using specific concentrator kits or precipitation protocols.
Plants also produce a variety of polysaccharides and secondary metabolites such as polyphenolic compounds and tannins (Leh et al., 2019). These compounds provide a challenge for RNA extraction as they can co-precipitate and thereby drastically decrease the purity and yield of RNA extractions from plants. Further, oxidized polyphenols or quinones can irreversibly bind to RNA causing damage to the molecules or inhibiting downstream reactions, such as reverse transcription used in NGS library preparation. Due to these challenges, researchers spend considerable time optimizing RNA extractions as the purity and quality of the RNA material directly corresponds to the resulting data quality. Classical methods, such as chloroform/phenol extraction, have shown clear advantages as they typically provide highly pure RNA. In addition, several commercial solutions exist with dedicated reagents designed to remove plant metabolites that may interfere with the extraction of subsequent reactions.
Classical Methods used to Extract RNA from Plants
Several different methods exist for extraction of RNA (and DNA) from plant material. As the challenges are vast and strongly dependent on the type of plant, tissue, and specific composition, these methods exist in various flavors. Protocols.io presents just one of the many resources that can be found online. Although the details of the individual steps may vary, the majority of publicly available protocols for RNA extraction from plants use one of the following methods:
- CTAB-based Extraction (Wang and Stegemann, 2010): The cetyltrimethylammonium bromide (or CTAB)-based extraction protocol is a widely spread method often used for plant material with high polysaccharide content and can be used for DNA as well as RNA. CTAB buffer typically consists of 2% cetyltrimethylammonium bromide, 1% polyvinylpyrrolidone, 100 mM Tris-HCl, 1.4 M NaCl, and 20 mM EDTA. Several variations and optimizations exist depending on the plant species or tissue from which RNA shall be extracted (Yan et al., 2022; Kiss et al., 2024).
Tipp: CTAB acts as a strong detergent and aids the disruption of the rigid cell wall. In addition, polyvinylpyrrolidone (PVP) – a component of CTAB buffer – complexes polysaccharide and polyphenol compounds. These are commonly found in plants and form precipitates with PVP when centrifuged. Afterwards the cleared lysate can be used for the further extraction steps and carry-over of these contaminants is thereby prevented. - Phenol-based Extraction (Box et al., 2011): Widely used classical RNA extractions for various difficult sample types, including tissues rich in proteins and lipids, rely on phenol. Phenol/chloroform-based extractions are also suitable for plants with high concentration of secondary metabolites, wax, or polysaccharides and deliver pure RNA at quite high yields compared to other methods. In the first step, the tissue is homogenized and cells are lysed in an RNA extraction buffer containing guanidinium salts. Subsequently, a mixture of phenol and chloroform is added. The extraction method then relies on phase separation of lysates based on their solubilization properties and subsequent selective isolation of molecules of interest. While nucleic acids will typically be dissolved in the aqueous phase, proteins are denatured and contained in the interphase, and lipids and other hydrophobic molecules will partition into the organic phase. This clear separation helps to remove contaminants and can greatly improve purity of the extracted nucleic acid.
Tipp: Visual inspection of the phase separation helps to ensure the success of the extraction. Precipitates may occur due to the protein and lipid content of the samples. If there are clear signs of precipitates in the aqueous phase, remove the phase and repeat the extraction with a renewed addition of phenol/chloroform.
Even though the use of phenol in labs can be challenging, the results are often of highest quality (Vennapusa et al., 2020). In addition, the use of acidic phenol can exclude DNA. Upon exposure, genomic DNA (gDNA) is also partitioned into the organic phase, leaving only RNA in the aqueous phase. This procedure can remove the need for additional enzymatic digests of co-isolated gDNA – not only minimizing the risk of contamination of RNA-based assays with DNA, but also preserving the integrity of the isolated RNA. While phenol/chloroform-based extraction methods excel in terms of RNA quality, contamination by carry-over of phenol and other organic solvents can also inhibit downstream reactions. The success of the method therefore depends also on the skill of the experimenter and how well the aqueous phase can be removed without touching the interphase or organic phase.
Tipp: Carry-over of organic solvents including phenol can be minimized by using phase separation aids. For example, Phase Lock Gel tubes included in the SPLIT RNA Extraction kit contain a gel that will create a physical barrier between the aqueous phase containing the RNA and the inter- and organic phase containing undesired molecules and organic solvents. Phase Lock Gel tubes therefore allow fast, comfortable, and safe phase separation without the risk of carry-over of contaminants. - Trizol-based Extraction: Trizol® is a commercial ready-made mix of guanidinium thiocyanate and phenol for the extraction of RNA (as well as DNA and protein) from various biological sample inputs. The principles of Trizol extraction are the same as for the phenol-based extraction discussed in the previous section.
Showcases for Classical Phenol-based RNA Extraction for Plant Science
Phenol/chloroform- based RNA extraction techniques are widely used in plant and crop sciences due to the high quality and purity of RNA that can be obtained. Often, the extraction is combined with a precipitation step which preserves RNA of all sizes, including small RNA, and maximizes total RNA yield. RNA extraction methods based on phenol have been described to be especially powerful in removing lipids, dealing with waxy outer surface layer, proteins, polysaccharides and polyphenols. For this reason several commercial kits utilizing this method are available to facilitate extraction and guarantee optimal composition of the reagents used, for example Trizol or Lexogen’s SPLIT RNA Extraction Kit.
The SPLIT RNA Extraction Kit enables fast and highly efficient extraction of high quality, high purity RNA from various plant tissues by efficient inactivation of RNases and removal of undesired secondary metabolites and enzyme inhibitors (Fig. 1). SPLIT uses Phase Lock Gel tubes for clear and easy phase separation minimizing the risk of phenol contamination. Thus, the obtained RNA is ideal for seamless NGS library preparation and other demanding applications such as full-length reverse transcription, sample preparation for microarray analysis, or RT-qPCR. Furthermore, SPLIT recovers the complete RNA size ranges, including small RNAs (≥17 nt) and is therefore also suitable in combination with small RNA analyses.
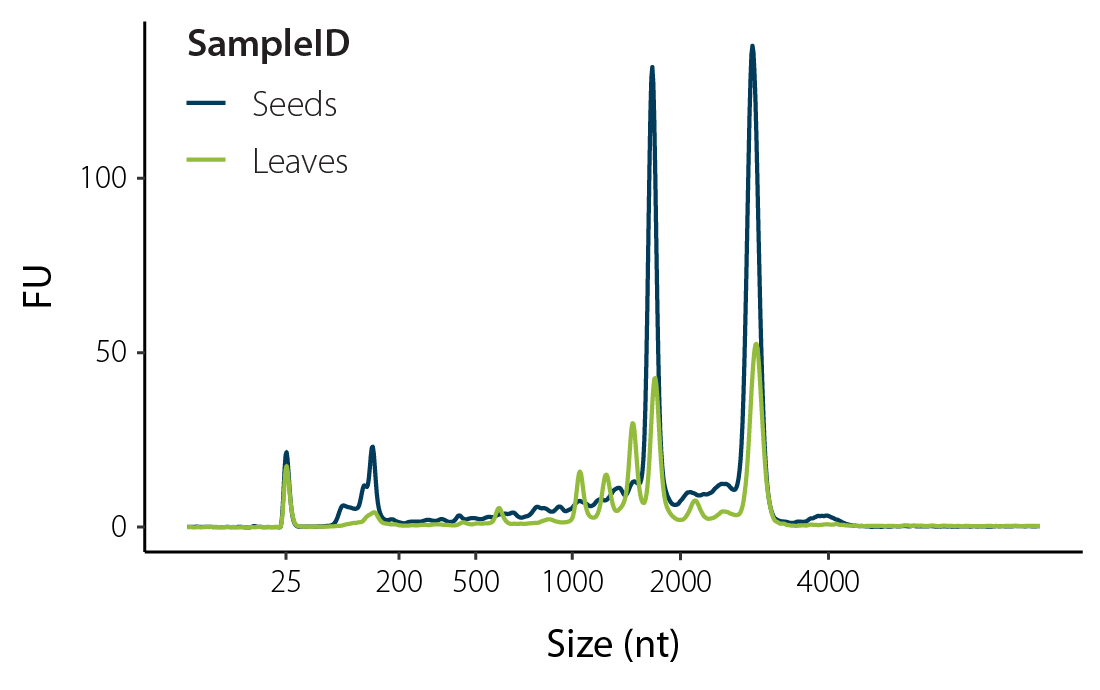
The following selected publications provide but a few examples of mechanisms commonly studied by analyzing plant RNA and showcase the use of SPLIT RNA Extraction. The three studies focus on transcriptional control in tomatoes, embryogenesis in barley, and immune responses in Arabidopsis.
Abstract
BSD (mammalian BTF2-like transcription factors, synapse-associated proteins, and DOS2-like proteins) is a conserved domain that exists in a variety of organisms, but its function has not been well studied. Here, we identified a novel BSD domain-containing protein (SlBSD1) in tomato (Solanum lycopersicum). Biochemical and microscopy assays indicated that SlBSD1 is a functional transcription factor that is predominantly localized in the nucleus. Loss-of-function and overexpression analyses suggested that SlBSD1 is a novel regulator of vegetative growth and leaf senescence in tomato. SlBSD1-knockdown (-KD) plants exhibited retarded vegetative growth and precocious leaf senescence, whereas SlBSD1-overexpression (-OX) plants displayed the opposite phenotypes. The negative role of SlBSD1 in leaf senescence was also supported by RNA-seq analysis comparing leaf tissues from SlBSD1-KD and wild-type plants. In addition, contents of soluble solids were altered in fruits in the SlBSD1-KD and SlBSD1-OX plants. Taken together, our data suggest that the novel transcription factor SlBSD1 plays important roles in controlling fruit quality and other physiological processes in tomato, including vegetative growth and leaf senescence.
Abstract
In barley, it is possible to induce embryogenesis in the haploid and uninucleate microspore to obtain a diploid plant that is perfectly homozygous. To change developmental fates in this fashion, microspores need to engage in cellular de-differentiation, interrupting the pollen formation, and restore totipotency prior to engaging in embryogenesis. In this work, we used the barley cultivar Gobernadora to characterize the transcriptome of microspores prior to (day 0) and immediately after (days 2 and 5) the application of a stress pretreatment. A deep RNA-seq analysis revealed that microspores at these three time points exhibit a transcriptome of ∼14k genes, ∼90% of which were shared. An expression analysis identified a total of 3,382 differentially expressed genes (DEGs); of these, 2,155 and 2,281 DEGs were respectively identified when contrasting expression at days 0 and 2 and at days 2 and 5. These define 8 expression profiles in which DEGs share a common up- or down-regulation at these time points. Up-regulation of numerous glutathione S-transferase and heat shock protein genes as well as down-regulation of ribosomal subunit protein genes was observed between days 0 and 2. The transition from microspores to developing embryos (days 2 vs. 5) was marked by the induction of transcription factor genes known to play important roles in early embryogenesis, numerous genes involved in hormone biosynthesis and plant hormonal signal transduction in addition to genes involved in secondary metabolism. This work sheds light on transcriptional changes accompanying an important developmental shift and provides candidate biomarkers for embryogenesis in barley.
Abstract
For over 60 years, salicylic acid (SA) has been known as a plant immune signal required for both basal and systemic acquired resistance (SAR). SA activates these immune responses by reprogramming up to 20% of the transcriptome through the function of NPR1. However, components in the NPR1-signaling hub, which appears as nuclear condensates, and the NPR1-signaling cascade remained elusive due to difficulties in studying transcriptional cofactors whose chromatin associations are often indirect and transient. To overcome this challenge, we applied TurboID to divulge the NPR1-proxiome, which detected almost all known NPR1-interactors as well as new components of transcription-related complexes. Testing of new components showed that chromatin remodeling and histone demethylation contribute to SA-induced resistance. Globally, NPR1-proxiome shares a striking similarity to GBPL3-proxiome involved in SA synthesis, except associated transcription factors (TFs), suggesting that common regulatory modules are recruited to reprogram specific transcriptomes by transcriptional cofactors, like NPR1, through binding to unique TFs. Stepwise greenCUT&RUN analyses showed that, upon SA-induction, NPR1 initiates the transcriptional cascade primarily through association with TGA TFs to induce expression of secondary TFs, predominantly WRKYs. WRKY54 and WRKY70 then play a major role in inducing immune-output genes without interacting with NPR1 at the chromatin. Moreover, a loss of NPR1 condensate formation decreases its chromatin-association and transcriptional activity, indicating the importance of condensates in organizing the NPR1-signaling hub and initiating the transcriptional cascade. This study demonstrates how combinatorial applications of TurboID and stepwise greenCUT&RUN transcend traditional genetic methods to globally map signaling hubs and transcriptional cascades.
Small RNA Isolation from Plant Samples
Small RNAs act as key regulators in diverse biological processes in eukaryotic organisms. Plants possess a vast repository of small RNA classes with diverse functions, for example microRNAs (miRNAs) and tasiRNAs (trans-acting small interfering RNAs) play important roles in plant growth and developmental processes such as embryogenesis, organ formation, shoot and root growth, and reproductive development. In addition, miRNAs and siRNA (small interfering RNAs) are key regulators orchestrating plant defense mechanisms upon exposure to pathogens and play a role in stress responses. Small RNAs therefore regulate fundamental processes and all aspects of plant life. To do so, they associate with specific proteins of the Argonaute family (AGOs) and form RNA-induced silencing complexes (RISCs) whereby the small RNAs guide the AGO proteins to their respective targets and elicit silencing of the specific target RNA.
To study small RNAs and their modus operandi, efficient protocols for small RNA isolation are necessary. As for extraction of total RNA described above, the composition of specific tissues and abundance of inhibitory molecules in plants presents a major challenge. Also in this case, phenol-based RNA extraction methods are commonly used.
Tipp: Combining phenol/chloroform extraction and clean-up by precipitation ensures that small RNAs are included in the final RNA eluate. When using column-based purification for small RNA experiments, use specific columns suited for sRNA purification and ensure that the size cutoff of the silica column does not exclude small RNAs. General columns may use a cutoff of 150 nt (small RNAs would be lost) while sRNA compatible columns retain molecules down to ~17 nt.
However, classical extraction does not resolve one of the major challenges in small RNA research: to determine which sRNAs are functional and which molecules are inactive artifacts picked up by the library preparation. Library generation for small RNA uses linker ligations to the 3’OH and 5′ phosphate moiety of the small RNA molecule. This ligation however, is not specific to small RNAs, but also incorporates all other RNA fragments in this respective size range into the library prep, for example fragmented tRNA, rRNA, or mRNA or even non-functional small RNA molecules. Not only precious sequencing reads are lost to these undesired by-products, it also presents a unique challenge to weed out the artifacts and determine which of the detected small RNAs are really active and functional.
Tipp: Small RNA library preps cover a range of sizes for molecules that can be ligated during library generation. Researchers can increase the efficiency of incorporating desired small RNAs by size selecting the fraction around 17 – 35 nt and thereby remove some of the larger tRNA or rRNA fragments prior to the library prep. However, while this method is widely used in research labs, it is quite tedious and time consuming as it requires RNA extraction from polyacrylamid gels and a lot of material is lost during the procedure due to inefficient recovery from the gel. Even though molecules of the right size can be enriched by this step, it does not guarantee that these small RNA are physiologically active and elicit a silencing function.
Isolation of Functional Small RNAs by Enrichment of RISCs
Small RNAs and other functional RNA fragments associate with Argonaute or AGO-proteins and assemble into RISCs to specifically silence their targets. This association of protein and sRNA has therefore been used by researchers to isolate functional small RNAs by purifying the AGO proteins, e.g., by co-immunoprecipitation (co-IP) prior to RNA extraction and analysis. While this highly sensitive method allows to isolate sRNA specifically associated with an AGO protein, the method itself is a tedious two-day procedure and is often limited by the availability of specific AGO-antibodies for the species or Argonaute protein of interest. In addition, many researchers are interested in non-model organisms with an unknown AGO repertoire and would need to first identify and characterize the AGO proteins, then purify them in sufficient amounts to generate antibodies before they can embark on the actual experiment.
TraPR (Trans-kingdom, rapid, affordable Purification of RISCs) was published in 2020 solving this dilemma for many researchers (Grentzinger et al., 2020). TraPR enables the specific isolation of functional sRNAs from RISCs. As a species-independent method, TraPR does not require any prior characterization of the sample or knowledge about the AGO proteins expressed in the plant.
TraPR Small RNA Isolation is a commercially available easy and robust 15-minute column purification method: the sample is lysed, and the clarified lysate is loaded onto the TraPR column. TraPR exploits the conserved properties of RISCs to elute them while bulk RNA and DNA are retained on the column (Fig 2).
After RISC elution, sRNAs can be isolated by e.g., phenol/chloroform extraction. The resulting pure sRNA fraction is suitable for all molecular biology and NGS applications, it can be directly used as input for small RNA library preps. Contaminating RNAs such as degradation products of tRNA, rRNA, and mRNA are effectively excluded from the purified RISC fraction allowing researchers to directly assess functional small RNAs and their regulatory networks.
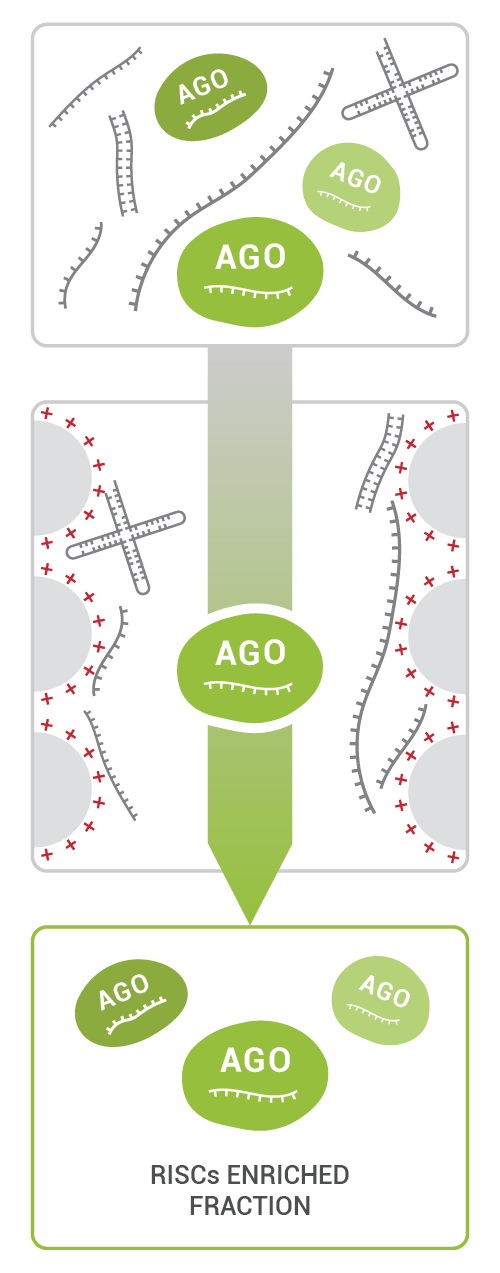
Tipp: The TraPR enrichment step is performed as ~15-minute procedure prior to RNA extraction and uses fresh plant lysate after homogenization as input. By using this small modification in the small RNA extraction protocol, researchers can eliminate the time-consuming polyacrylamid gel size selection step to enrich RNAs between 17 – 35 nt in length prior to library generation. The exclusion of degradation products during the TraPR procedure results in more sequencing reads mapping to small RNAs ensuring researchers obtain meaningful data for subsequent analysis.
Comparisons between small RNA profiles generated from TraPR small RNA extractions vs. small RNA-Seq from total RNA preparations allow to distinguish physiologically active small RNAs, active small RNA isoforms and sRNA modifications within the complete sRNA repertoire of an organism (Dallaire et al., 2021). These insights are particularly useful for plants as they possess a vast amount of small RNAs with often unknown functions. TraPR can help characterize these key regulators in different plant species and link them to important physiological processes of agricultural relevance. In the next section, we provide several examples of how TraPR combined with small RNA sequencing has been used in important crops.
Showcases for Functional Small RNA Isolation for Important Plants and Crops
The highlighted publications in the following section showcase the advantages of functional small RNA enrichment to unravel regulatory circuits. In the studies shown below, researchers focus on the generation of microRNA profiles controlling dormancy, the characterization of fertility-related phasiRNAs in Zea mays and related species, the control of polyploidy-related paramutation by small RNAs in Arabidopsis, and transcriptional and epigenetic regulation of the important plant symbiont Rhizophagus.
Abstract
Winter dormancy is an adaptative mechanism that temperate and boreal trees have developed to protect their meristems against low temperatures. In apple trees (Malus domestica), cold temperatures induce bud dormancy at the end of summer/beginning of the fall. Apple buds stay dormant during winter until they are exposed to a period of cold, after which they can resume growth (budbreak) and initiate flowering in response to warmer temperatures in spring. It is well-known that small RNAs modulate temperature responses in many plant species, but however, how small RNAs are involved in genetic networks of temperature-mediated dormancy control in fruit tree species remains unclear. Here, we have made use of a recently developed ARGONAUTE (AGO)-purification technique to isolate small RNAs from apple buds. A small RNA-seq experiment resulted in the identification of 17 micro RNAs (miRNAs) that change their pattern of expression in apple buds during dormancy. Furthermore, the functional analysis of their predicted target genes suggests a main role of the 17 miRNAs in phenylpropanoid biosynthesis, gene regulation, plant development and growth, and response to stimulus. Finally, we studied the conservation of the Arabidopsis thaliana regulatory miR159-MYB module in apple in the context of the plant hormone abscisic acid homeostasis.
Abstract
Reproductive phasiRNAs (phased, small interfering RNAs) are broadly present in angiosperms and play crucial roles in sustaining male fertility. While the premeiotic 21-nt (nucleotides) phasiRNAs and meiotic 24-nt phasiRNA pathways have been extensively studied in maize (Zea mays) and rice (Oryza sativa), a third putative category of reproductive phasiRNAs–named premeiotic 24-nt phasiRNAs–have recently been reported in barley (Hordeum vulgare) and wheat (Triticum aestivum). To determine whether premeiotic 24-nt phasiRNAs are also present in maize and related species and begin to characterize their biogenesis and function, we performed a comparative transcriptome and degradome analysis of premeiotic and meiotic anthers from five maize inbred lines and three teosinte species/subspecies. Our data indicate that a substantial subset of the 24-nt phasiRNA loci in maize and teosinte are already highly expressed at the premeiotic phase. The premeiotic 24-nt phasiRNAs are similar to meiotic 24-nt phasiRNAs in genomic origin and dependence on DCL5 (Dicer-like 5) for biogenesis, however, premeiotic 24-nt phasiRNAs are unique in that they are likely i) not triggered by microRNAs, ii) not loaded by AGO18 proteins, and iii) not capable of mediating PHAS precursor cleavage. In addition, we also observed a group of premeiotic 24-nt phasiRNAs in rice using previously published data. Together, our results indicate that the premeiotic 24-nt phasiRNAs constitute a unique class of reproductive phasiRNAs and are present more broadly in the grass family (Poaceae) than previously known.
Abstract
Paramutation is a form of non-Mendelian inheritance in which the expression of a paramutable allele changes when it encounters a paramutagenic allele. This change in expression of the paramutable alleles is stably inherited even after segregation of both alleles. While the discovery of paramutation and studies of its underlying mechanism were made with alleles that change plant pigmentation, paramutation-like phenomena are known to modulate the expression of other traits and in other eukaryotes, and many cases have probably gone undetected. It is likely that epigenetic mechanisms are responsible for the phenomenon, as paramutation forms epialleles, genes with identical sequences but different expression states. This could account for the intergenerational inheritance of the paramutated allele, providing profound evidence that triggered epigenetic changes can be maintained over generations. Here, we use a case of paramutation that affects a transgenic selection reporter gene in tetraploid Arabidopsis thaliana. Our data suggest that different types of small RNA are derived from paramutable and paramutagenic epialleles. In addition, deletion of a repeat within the epiallele changes its paramutability. Further, the temperature during the growth of the epiallelic hybrids determines the degree and timing of the allelic interaction. The data further make it plausible why paramutation in this system becomes evident only in the segregating F2 population of tetraploid plants containing both epialleles. In summary, the results support a model for polyploidy-associated paramutation, with similarities as well as distinctions from other cases of paramutation.
Abstract
Arbuscular mycorrhizal (AM) fungi form mutualistic relationships with most land plant species. AM fungi have long been considered as ancient asexuals. Long-term clonal evolution would be remarkable for a eukaryotic lineage and suggests the importance of alternative mechanisms to promote genetic variability facilitating adaptation. Here, we assessed the potential of transposable elements for generating such genomic diversity. The dynamic expression of TEs during Rhizophagus irregularis spore development suggests ongoing TE activity. We find Mutator-like elements located near genes belonging to highly expanded gene families. Whole-genome epigenomic profiling of R. irregularis provides direct evidence of DNA methylation and small RNA production occurring at TE loci. Our results support a model in which TE activity shapes the genome, while DNA methylation and small RNA–mediated silencing keep their overproliferation in check. We propose that a well-controlled TE activity directly contributes to genome evolution in AM fungi.
References
Box, M.S., Coustham, V., Dean, C., and Mylne (2011) Protocol: A simple phenol-based method for 96-well extraction of high quality RNA from Arabidopsis. Plant Methods 7. doi: 10.1186/1746-4811-7-7
Dallaire A., Manley B. F., Wilkens, M., Bista, I., Quan, C., Evangelisti, E., Bradshaw, C. R., Ramakrishna, N. B., Schornack, S., Butter, F., Paszkowski, U., and Miska, E.A. (2021) Transcriptional activity and epigenetic regulation of transposable elements in the symbiotic fungus Rhizophagus irregularis. Genome Res. 31:2290-2302, doi: 10.1101/gr.275752.121
Grentzinger, T., Oberlin, S., Schott, G., Handler, D., Svozil, J., Barragan-Borrero, V., Humbert, A., Duharcourt, S., Brennecke, J., and Voinnet O. (2020) A universal method for the rapid isolation of all known classes of functional silencing small RNAs, Nucleic Acids Res. 48:e79, doi: 10.1093/nar/gkaa472
Kiss, T., Karácsony, Z., Gomba-Tóth, A. Szabadi, K.L., Spitzmüller, Z., Hegyi-Kaló, J., Cels, T., Otto, M., Golen, R., Hegyi, A. I., Geml, J., and Váczy., K. Z. (2024) A modified CTAB method for the extraction of high-quality RNA from mono-and dicotyledonous plants rich in secondary metabolites. Plant Methods 20, 62. doi: 10.1186/s13007-024-01198-z
Leh T.Y., Yong C.S.Y., Nulit R., Abdullah J.O. (2019) Efficient and High-Quality RNA Isolation from Metabolite-Rich Tissues of Stevia rebaudiana, an Important Commercial Crop. Trop Life Sci Res. 30:149-159, doi: 10.21315/tlsr2019.30.1.9
Vennapusa, A.R., Somayanda, I.M., Doherty, C.J., Jagadish, S. V. K. (2020) A universal method for high-quality RNA extraction from plant tissues rich in starch, proteins and fiber. Sci Rep 10, 16887 (2020). doi: 10.1038/s41598-020-73958-5
Wang L., and Stegemann J.P. (2010) Extraction of high quality RNA from polysaccharide matrices using cetyltrimethylammonium bromide. Biomaterials. 31:1612-8. doi: 10.1016/j.biomaterials.2009.11.024
Yan, W.J., Pendi, F.H. and Hussain, H. (2022) Improved CTAB method for RNA extraction of thick waxy leaf tissues from sago palm (Metroxylon sagu Rottb.). Chem. Biol. Technol. Agric. 9:63, doi: 10.1186/s40538-022-00329-9
Written by Dr. Yvonne Goepel







